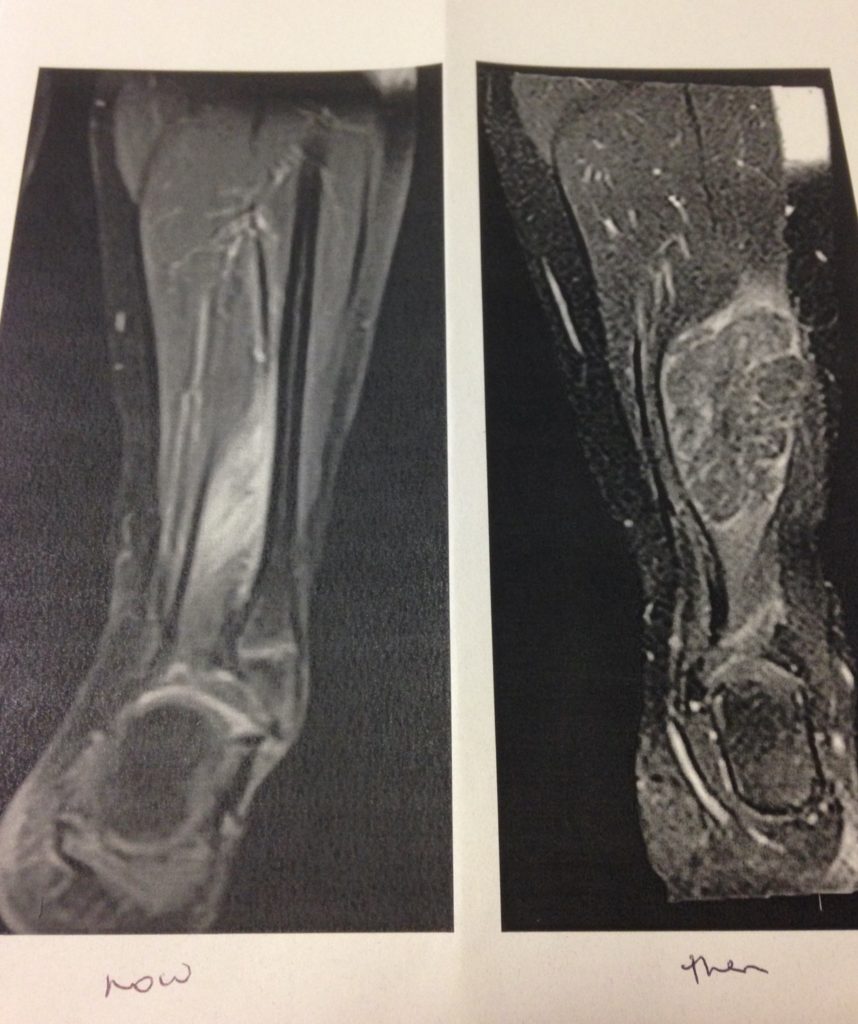
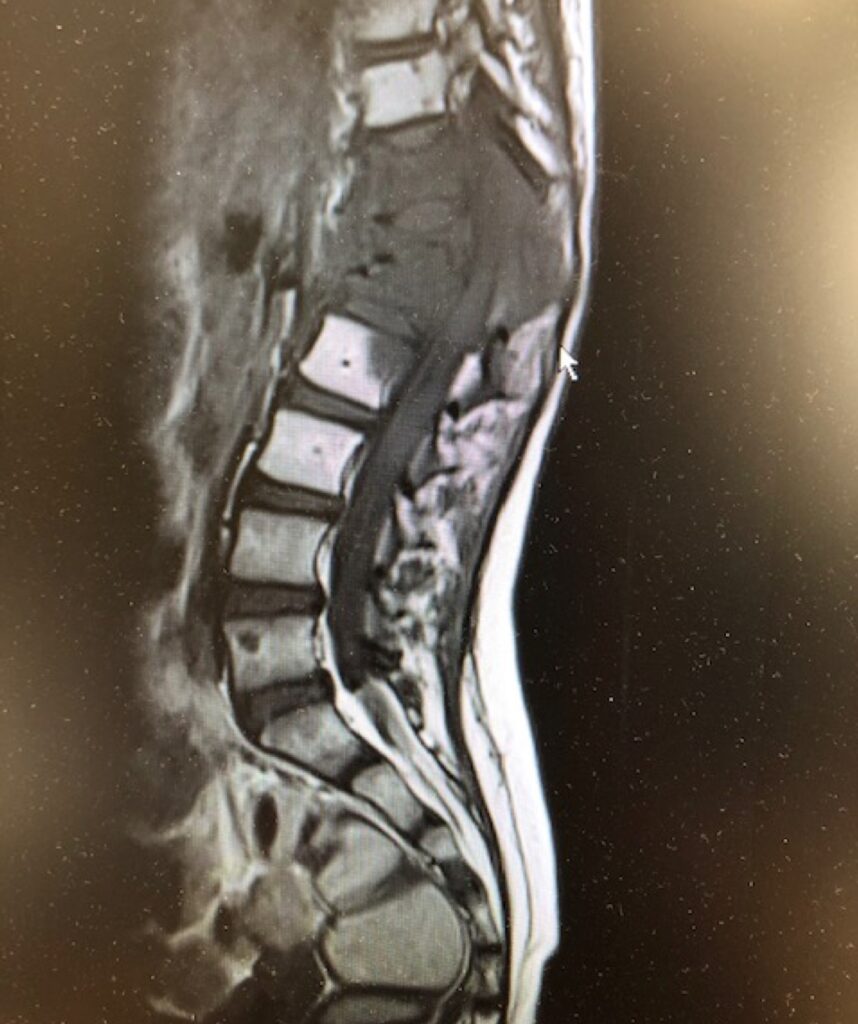
Diagnosis and prognosis
There are different types and the prognosis varies a lot depending on which type of rhabdomyosarcoma the patient has.
Once you have a diagnosis, rhabdo may be shortened to ARMS (alveolar), ERMS (embryonal), or another abbreviation ending RMS, prefixed by letters indicating the type, location, or both.
Research has highlighted the importance of ‘fusion testing’ in addition to histological classification at diagnosis so that patients can be allocated to appropriate low, intermediate or high risk groups and their treatment tailored appropriately.
Read this paper for more information about the complexities of rhabdomyosarcoma diagnosis. The summary states that ‘poor outcomes associated with ARMS likely relates to the presence of PAX-FOXO1 fusion genes and not to histological features’.
This is why research is so vital for this disease; there is still a lot to understand and effective medicines to find. There is currently no known cure for relapsed metastatic alveolar rhabdomyosarcoma.
How common is it?
8% of childhood deaths in Scotland from 2014 to 2019 were cancer related.
Rhabdomyosarcoma accounts for 5% of the childhood cancer deaths in Scotland during the same period.
This means that 0.4% of childhood deaths in Scotland are due to rhabdomyosarcoma. It is rare. It is considered an ‘orphan’ disease.
Because it’s rare, it gets little publicity, little funding and little progress is therefore made to change the prognosis for children diagnosed with the worst forms.
How is it treated?
Standard treatment consists of chemotherapy, surgery and radiotherapy. Some patients may receive brachytherapy and/or proton beam therapy.